Modeling with PyRy3D
- 1. Why to use PyRy3D?
- 2. What are the advantages of using PyRy3D?
- 3. Modeling of flexible and disordered regions with PyRy3D
- 4. How to analyze models obtained from PyRy3D?
- 5. Predicting accuracy of PyRy3D models
- 6. Refinement of low-resolution models generated by PyRy3D
- 7. How PyRy3D will be developed in the future?
1. Why to use PyRy3D?
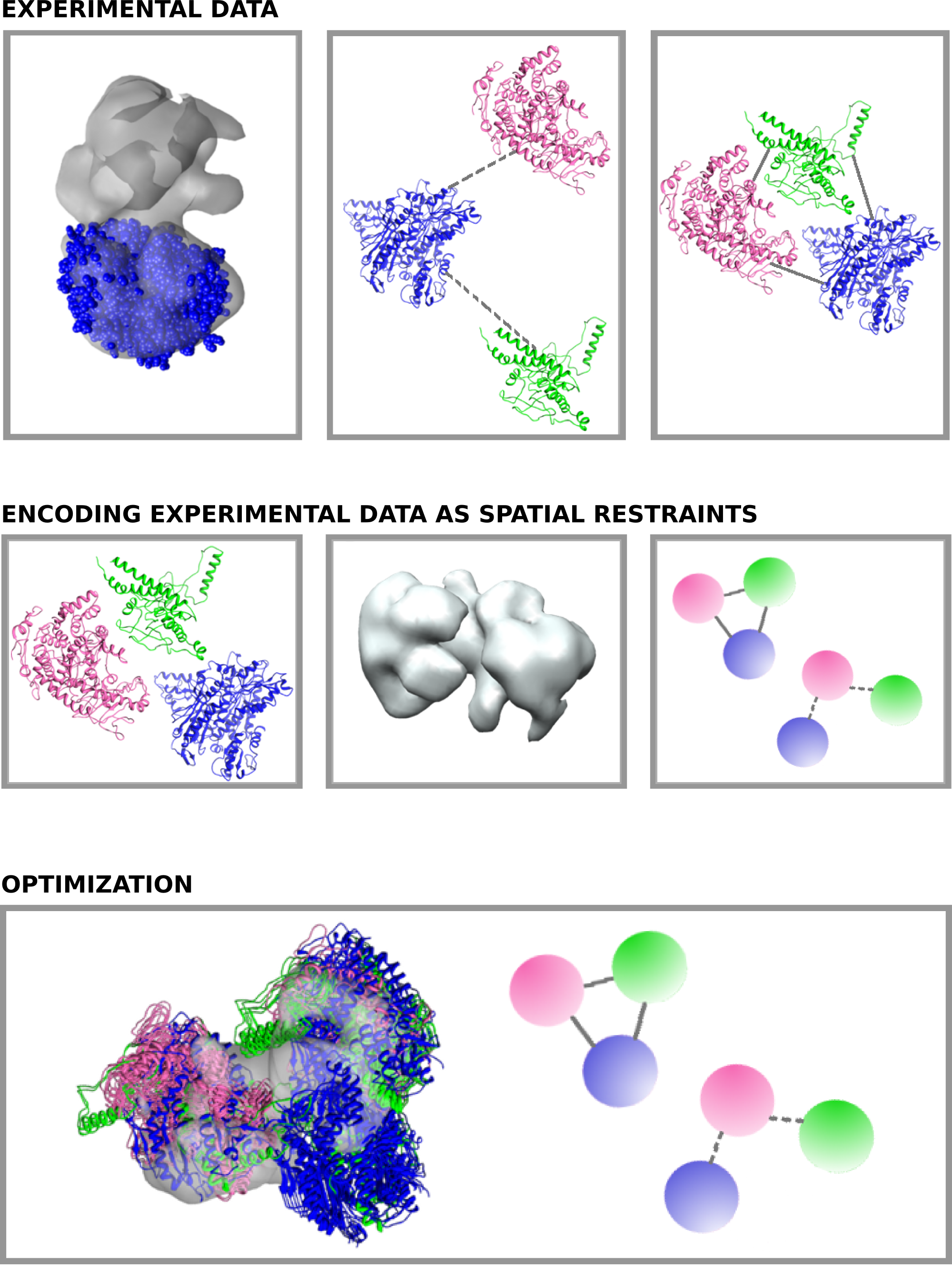
PyRy3D is a novel bioinformatic tool dedicated to model large macromolecular complexes. It performs superbly when compared to other available methods (such as Situs and Multifit server) for fitting atomic structures assemblies into electron density maps. Additionally, PyRy3D is a user-friendly, ready-to-use software for performing hybrid modeling by incorporating both experimental and predicted data.
PyRy3D program was classified as one of the top three finalists during The ISMB 2011 Killer Application Award Competition, where a committee selects tools or systems of most practical benefit to biochemists and/or molecular biologists. To be eligible for the Killer Application Award, a system or a tool must also be fully functional, and be presented at the ISMB conference. Several scientists in their modelling tasks have already successfully used PyRy3D, and some of their opinions about the program can be found here.
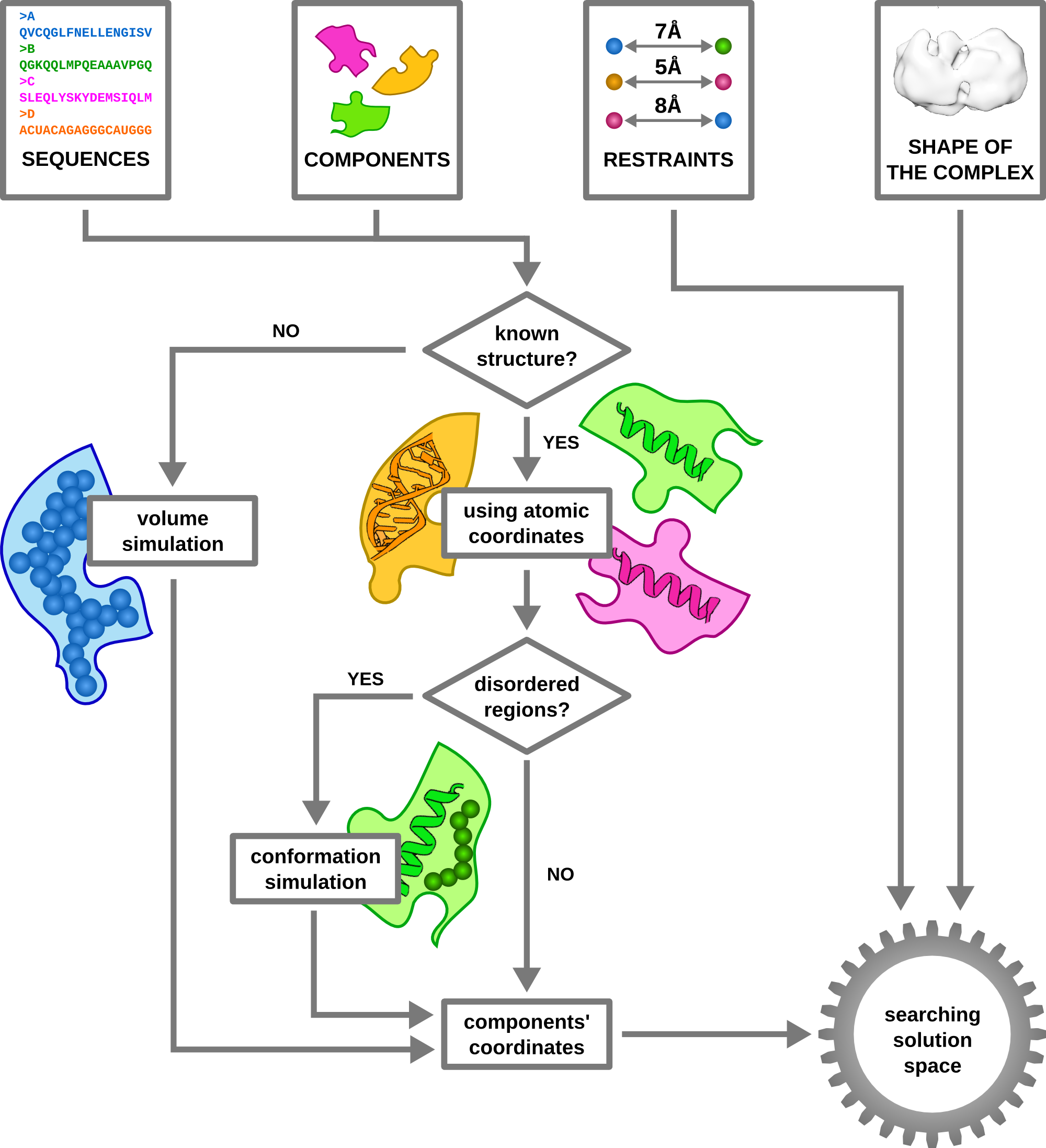
2. What are the advantages of using PyRy3D?
- can work with wide range of input data types
As an integrative method, PyRy3D works with a wide variety of data, such as structures, distance restraints, information about solvent exposure and complex volume. It can also work with components containing disordered fragments, or no tertiary structure at all.
- is well documented
PyRy3D is a product ready to use with full documentation, user-friendly GUI compatible with UCSF Chimera software package and the online server.
- is available as a standalone and server version
PyRy3D can be run in two modes: "automatic", where a user provides input data and simulation parameters and receives models fulfilling restraints (via command-line application or server), but also, thanks to the GUI, as a "fully controlled tool" where visualization of simulation and interpretation of program results is possible.
- allows to modify parameters easily
We put a lot of effort in providing the users with the possibility to modify all program parameters for full control over the modeling process. For example, users can easily change simulation algorithm (genetic, simulated annealing, replica exchange), weights of scoring function elements, mutation types and their frequencies. Users can also freeze components inside an electron density map, or define their movements' limitations. Also, thanks to the unique scaling system implemented in the program, one can easily set modification of values of particular parameters during simulation (e.g. by introducing low penalties for clashes at the beginning, and very high at the end). Usage of all of the aforementioned features is simple and requires only minor changes in a text file (configuration file), or a selection of particular options in the GUI.
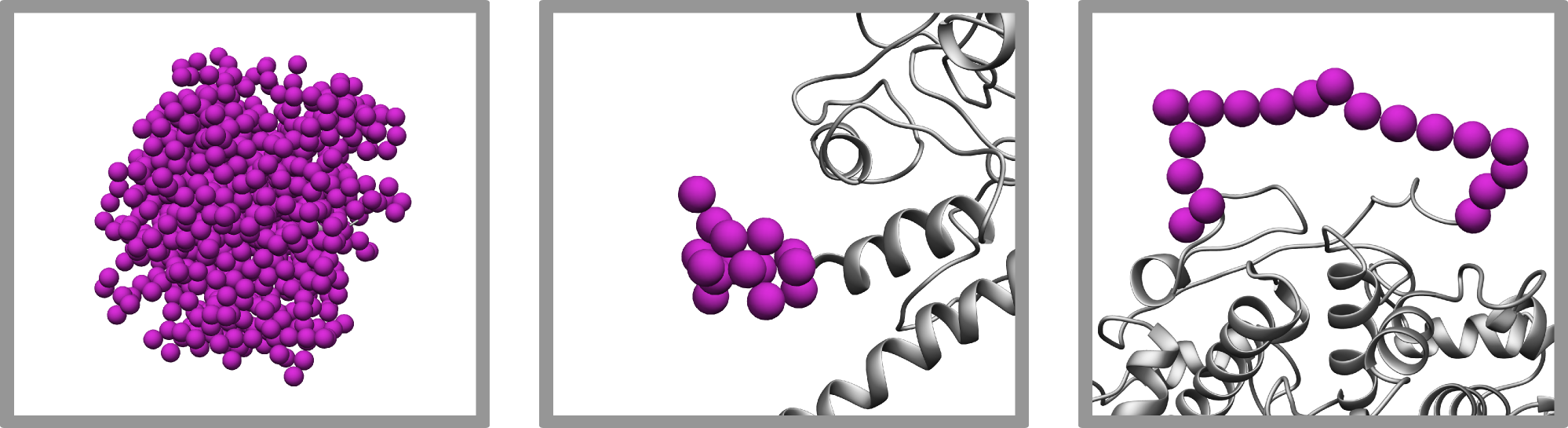
3. Modeling of flexible and disordered regions with PyRy3D
A unique feature of PyRy3D software is modeling of complexes with flexible or disordered fragments and even for sequences without a defined structure at all. During the complex building procedure, PyRy3D changes conformations of these regions (or their shape) to mimic their dynamics. Owing to this feature, PyRy3D enables to predict very low resolution models even for complexes where 3D structures of some components are unknown or highly disordered. However, the flexibility modeling implemented in our method enables to fill the density map shape by adequate complex volume rather than building realistic full-atom models. For example, all simulated regions are composed of pseudoatoms of a given radius (3.5A for proteins and 7.5A for nucleic acids). As a consequence, all fragments added by PyRy3D should be reconstructed into full-atom representation. For this purpose we recommend programs for loops modeling, such as Mod-EM.
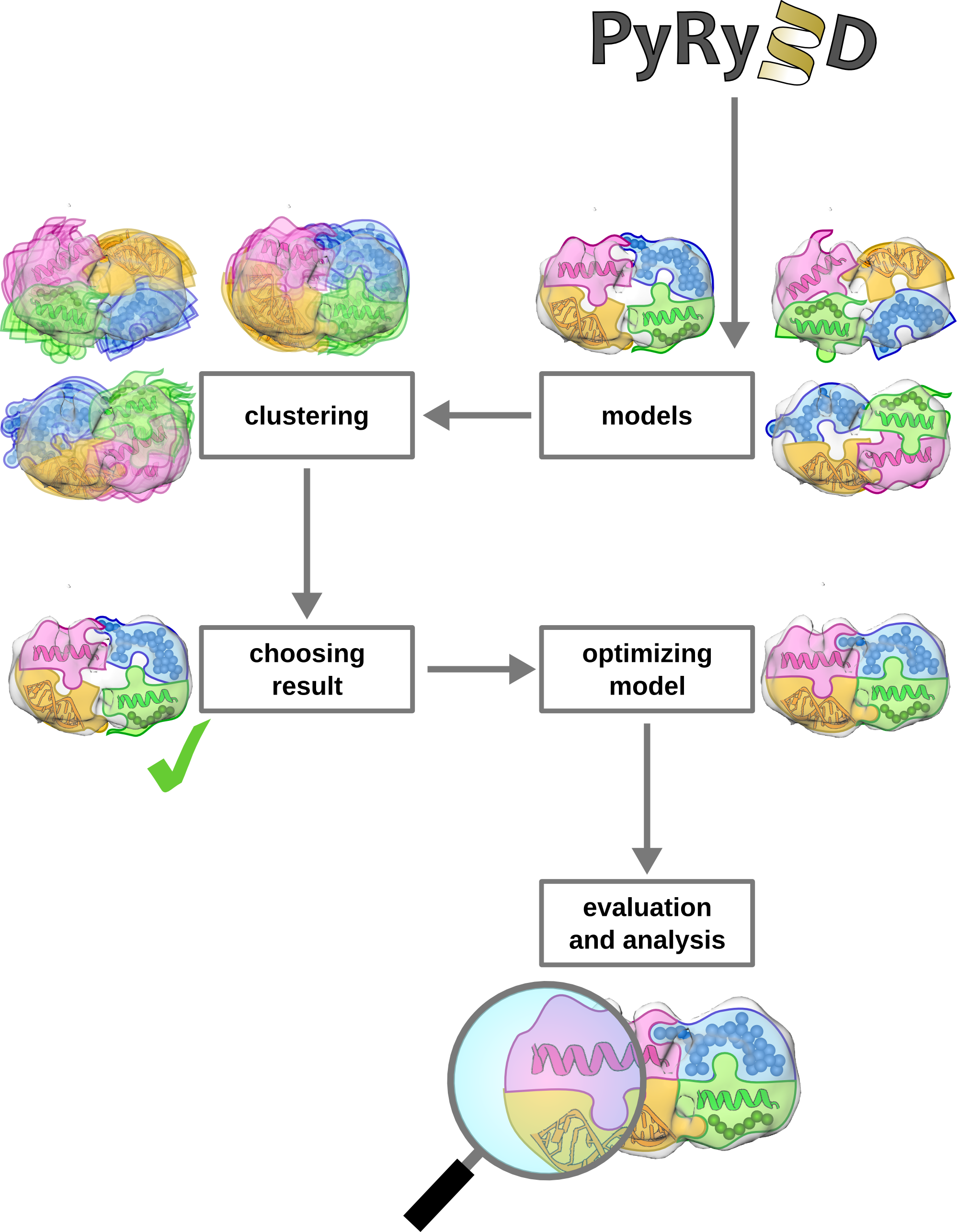
4. How to analyze models obtained from PyRy3D?
A single run of the program generates a trajectory of models (usually around 100 000). Since PyRy3D applies Monte Carlo algorithm, we strongly recommend to perform many independent runs of the program (100 minimum). Then, to check the consistency of generated models, clustering method should be applied for models with highest scores obtained from independent runs. Clustering tool is distributed with PyRy3D package and can be run as a simple python script via command line or via GUI.
Obtaining one cluster of solutions means that there is only one possible solution and an obtained model can be used as a starting point to infer structure-function relationships. Two or more clusters suggest that data used for modelling is insufficient to predict complex structure or a program identified multiple conformational states of the complex. In such cases closer verification of data used for modeling and/or performing additional experiments to retrieve more accurate data is necessary.
5. Predicting accuracy of PyRy3D models
Assessing the accuracy of computational models is an essential step in structure prediction process and all models should be verified. The simplest method, is to verify a fulfilment of experimental restraints used for modelling in obtained models. In order to check data consistency we recoomend Filtrest3D software. It allows for discrimination of a large number of alternative models of structures against a set of restraints derived from low-resolution experimental analyses. However, bear in mind that PyRy3D generates models already fulfilling input restraints. Thus far, we advise to validate obtained models against an independent set of restraints (e.g. data not used for modeling).
Also, the similarity of generated models satisfying input restraints indicates consistency of obtained results. For this purpose, clustering procedures can be applied, such as MaxCluster, MM-align or ROSSETA. PyRy3D also provides a clustering tool that uses algorithm similar to that implemented originally in ROSETTA. Among clusters, statistically most probable model should be classified as the best scored representative from the largest cluster of solutions.
Last but not least, very important step in model evaluation is a visual inspection of generated models. This particular type of assessment depends on research objective and one can use different features to verify obtained models, such as binding site exposure, electrostatic potential or interactions between components.
All of the aforementioned quality assessment procedures can also be performed via Python scripts distributed with PyRy3D package, and via easy-to-use tools in PyRy3D Chimera Extension. With the use of a graphical interface, a user can visualize alternative models based on a set of restraints, investigate CCC values for models docked into density maps, cluster models based on RMSD or CCC values, as well as inspect clashes.
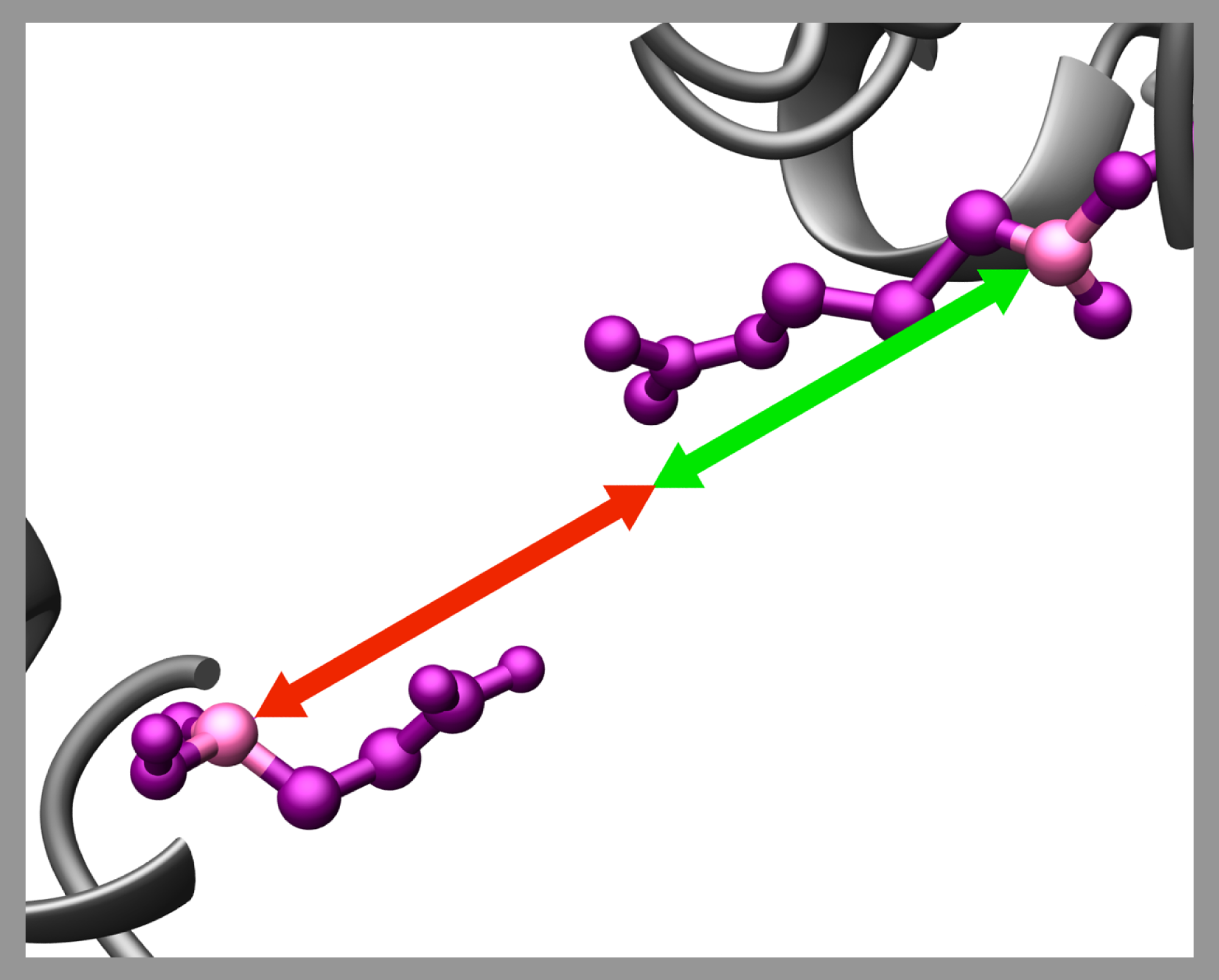
6. Refinement of low-resolution models generated by PyRy3D
Models generated by PyRy3D are of low resolution and they always should be verified and optimized. The improvement includes refinement of local fit to an electron density map, removing clashes between side chains or building full atom models for flexible/disordered fragments inserted by PyRy3D.
First, to improve local fit of components into electron density map, tools such as DEFINER or colores can be used.
Also, since procedures for inserting missing fragments into structures in PyRy3D are rather stochastic, we recommend to rebuild them with loop modeling methods such as Refiner, Superlooper, ROSETTA or ModLoop or apply flexible docking programs, such as Mod-EM, Moulder-EM or qplasty.
Finally, clashes and chain distortions can be repaired with homology modelling programs such as Modeller and SWISS-MODEL (for proteins), and ModeRNA or MacroMoleculeBuilder (for RNA), as well as molecular dynamics procedures implemented in NAMD or Zephyr.
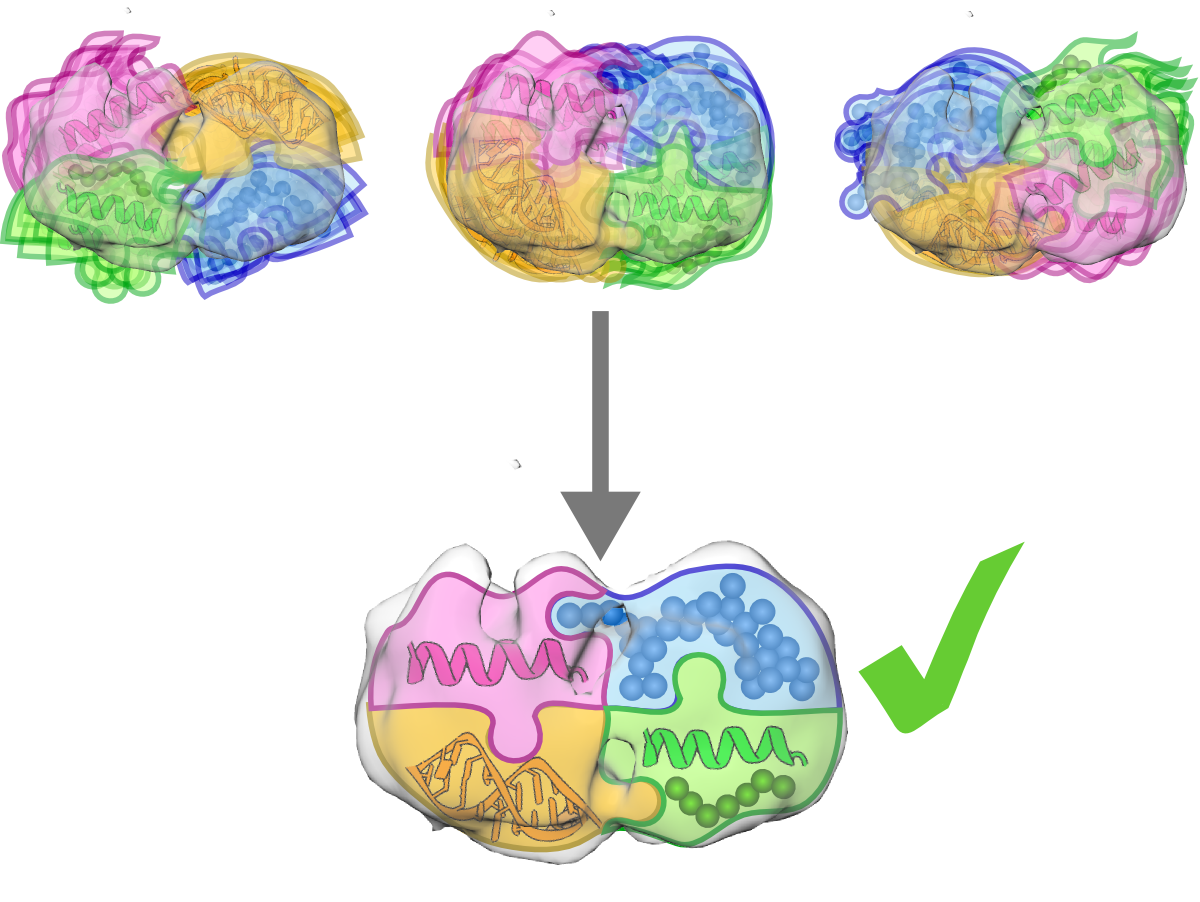
7. How will PyRy3D be developed in the future?
In the future versions of the program we will focus on implementation of new restraint types (such as excluded volume, secondary structure, angles etc.) and introduction of raw data from Small Angle Scattering as complex shape descriptors. Also, we would like to impose conformational changes of flexible and disordered regions by inserting fragments derived from X-ray structures into models instead of random positioning of pseudoatoms.
Moreover, the scoring function will be enriched by the factor corresponding to the evaluation of interactions within complex components. In particular, we intend to implement coarse-grained potentials that approximate energies of protein-protein and protein-nucleic acid interactions such as DARS-RNP or DECK.
Finally, we would like to incorporate local optimization procedures, such as those implemented in MultiFit (DOMINO), Situs or MinkoFit3D (DEFINER), to improve local fit of models into electron density maps. However, we will create optimization method that refines local fit of structures into density maps without violating user-defined restraints.