
| | |||||||||||||||||
| |||||||||||||||||
Secondary structure in ModeRNA
- Introduction
- Calculating 2D structures
- Making a helix longer
- Making a helix shorter
- Adding Watson-Crick paired bases
- Finding linkers with given 2D structure
- Adding custom 2D elements
Introduction
Secondary (2D) structure includes typical Watson-Crick pairing as well as GU wobble pairs. ModeRNA provides a set of functions that can be applied to specify 2D structure in the model, in particular in regions where the backbone conformation is modified (insertions and deletions). 2D structure restraints for fragment search can be provided by the user in the Vienna format.
Base pairs in helical conformation can be added to the model (e.g. to extend existing helices or insert new ones), For unpaired bases, complementary bases can be added (e.g. to build a helix starting from an unpaired single strand). Entire fragments with defined 3D coordinates (provided by the user) can be inserted into the model. Fragments can be also removed. A detailed description of all functions for secondary structure modeling is presented below.
In all pictures the following code is used:

Calculating 2D structures
get_secstruc(structure)
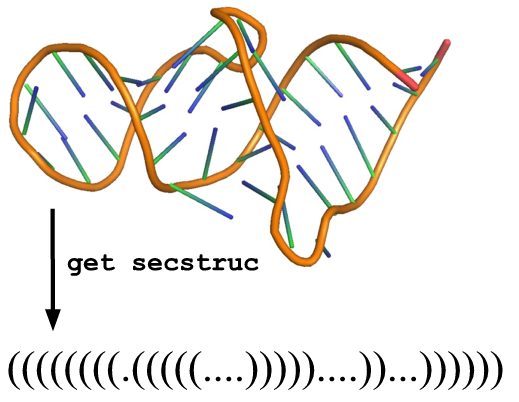
Description:
Extract secondary structure from a given 3D structure.
Arguments:
structure (model or template or any other ModernaStructure object)
Example
[ download template ]from moderna import *
t = load_template('1F1T.pdb', 'A')
clean_structure(t)
print get_secstruc(t)
Making a helix longer
extend_helix(model, anchor5, anchor3, new_sequence)
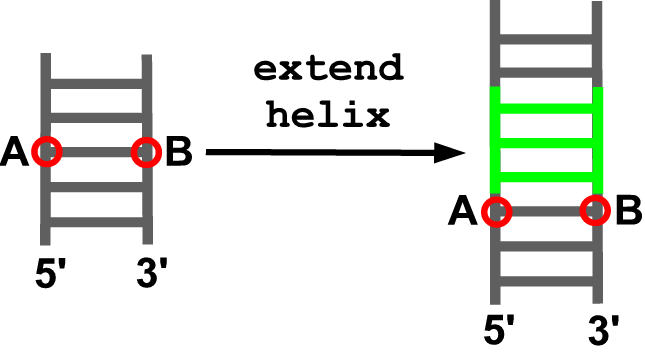
Description
This function extends a helix. A helical fragment (default: moderna/data/helix.pdb) is added as to generate an extension of the indicated anchoring residues in the model. In case a part of the model is between the indicated anchoring residues it is automatically found and moved as to become anchored to the end of the inserted fragment.
Arguments
- model object
- identifier of the anchoring residue at the 5’ end of the insertion site (A)
- identifier of the anchoring residue at the 3’ end of the insertion site (B)
- sequence of the helical fragment to be added (starting from the 5’ end of the insertion site with the strands separated by an underscore)
Example 1
[ download template ] [ download alignment ]from moderna import *
t = load_template('1F1T.pdb', 'A')
clean_structure(t)
a = load_alignment('alignment_1F1T.fasta')
m = create_model()
apply_alignment(t, a, m)
extend_helix(m, '2', '37', Sequence('CG_CG'))
Example 2
[ download template ] [ download alignment ]from moderna import *
t = load_template('1KXK.pdb', 'A')
clean_structure(t)
a = load_alignment('alignment_1KXK.fasta')
m = create_model()
apply_alignment(t, a, m)
extend_helix(m,'22','51',Sequence('AUG_CAU'))
Example 3
[ download template ] [ download alignment ]from moderna import *
t = load_template('1XJR.pdb', 'A')
clean_structure(t)
a = load_alignment('alignment_1XJR_long.fasta')
m = create_model()
apply_alignment(t, a, m)
extend_helix(m,'5','45',Sequence('GCGCGC_GCGCGC'))
Making a helix shorter
shrink_helix(model, anchor5, anchor3, anchor5_up, anchor3_up)
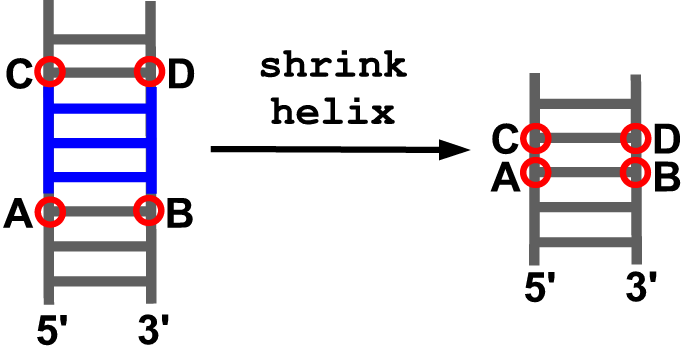
Description
Removes a fragment of an existing helix between the indicated anchoring residues. Afterwards both remaining parts are connected.
Arguments
- model object
- anchoring residue at the 5’ end of the insertion site (A)
- anchoring residue at the 3’ end of the insertion site (B)
- anchoring residue in the downstream part of the 5’ strand (C)
- anchoring residue in the upstream part of the 3’ strand (D)
Example
[ download template ]# eliminates a few base pairs from a helix.
m = load_model('hairpin_14nt.pdb', 'R')
shrink_helix(m, '2', '13', '6', '10')
fix_backbone(m)
Adding Watson-Crick paired bases
add_pair_to_base(model, anchor, new_id, new_sequence)
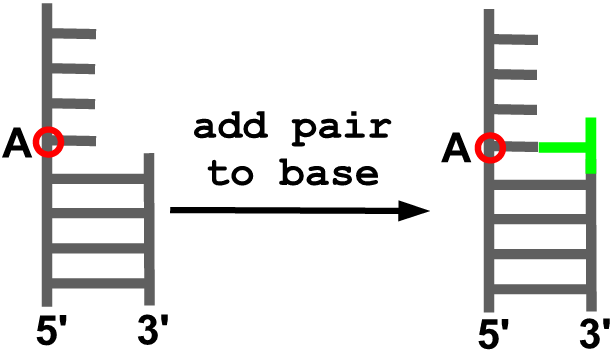
Description
Adds a residue to form a Watson-Crick pair with the indicated residue in the model.
Arguments
- model object
- residue to which a pair will be added (A)
- identifier (residue number) for the residue to be added
- residue type for the new residue [A, U, G, C]
Example
[ download template ]# adds three base pairs to a single strand.
m = load_model('single_strand.pdb')
add_pair_to_base(m, '1', '20', Sequence('C'))
add_pair_to_base(m, '2', '19', Sequence('C'))
add_pair_to_base(m, '3', '18', Sequence('C'))
# glue the new residues together
fix_backbone(m,'18','19')
fix_backbone(m,'19','20')
Finding linkers with given 2D structure
find_loop(model, anchor5, anchor3, sequence, number_of_candidates, 2D_struc)and
insert_fragment(model, fragment)
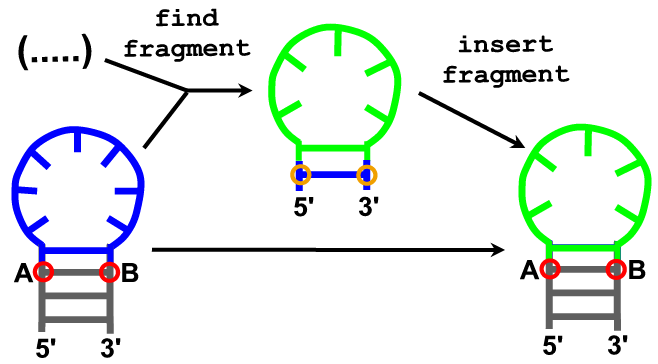
Description
Specification of 2D structure can be used to restrict fragment selection in the fragment insertion procedure.
Arguments (find_loop)
- model
- anchoring residue at the 5’ end of the insertion site. (A)
- anchoring residue from the 3’ end of the insertion site. (B)
- sequence
- number of fragment candidates that will be reported (or all that are found).
- secondary structure in dot-bracket notation.
Arguments (insert fragment)
- model
- fragment
Example
[ download template ]# retrieves a haipin loop from the linker database.
m = load_model('hairpin_14nt.pdb', 'R')
print m.get_sequence()
print m.get_secstruc()
candidates = find_fragment(m, '6', '10', Sequence('GGGGUUUUCCCC'), 10, secstruc='((((....))))')
print candidates[0]
print candidates[0].fragment_instance.get_secstruc()
insert_fragment(m, candidates[0])
Adding custom 2D elements
insert_2D_fragment(model, anchor5, anchor3, anchor5_up, anchor3_up, anchor5_fr, anchor3_fr, anchor5_up_fr, anchor_3_up_fr, fragment_file, chain_name)
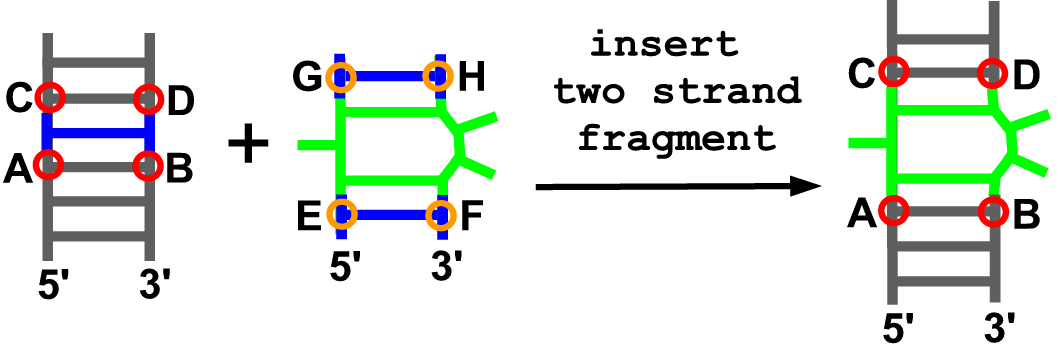
Description
With this function users can add their own 3D fragment containing two strands to an existing model. This operation requires PDB-formatted files with coordinates of the model to be modified and the fragment to be inserted, and the definition of anchoring residues: four from the model and four from the fragment.
Arguments
- model
- anchoring residue at the 5’ end of the model (A)
- anchoring residue at the 3’ end of the model (B)
- anchoring residue at the upper part on 5’ strand of the model (C)
- anchoring residue at the upper part on 3’ strand of the model (D)
- anchoring residue at the 5’ end of the fragment (E)
- anchoring residue at the 3’ end of the fragment (F)
- anchoring residue at the upper part on 5’ strand of the fragment (G)
- anchoring residue at the upper part on 3’ strand of the fragment (H)
- pdb file with coordinates of the fragment
- chain ID of given fragment
Example
[ download model ] [ download bulge fragment ]# adds a single-nucleotide bulge between some residues of the hairpin.
m = load_model('hairpin.pdb', 'D')
insert_two_strand_fragment(m, '30', '40', '31', '39', '195', '219', '196', '217', 'bulge.pdb','A')
|
| Copyright© - Adam Mickiewicz University - All rights reserved |