Tutorial
Introduction
This tutorial is a very brief introduction to the capabilities of the PyRy3D software tool – the program for modeling of structures for large macromolecular complexes.
Particularly you will learn:
- how to prepare input data about your systems for PyRy3D simulation
- how to manipulate with simulation parameters
- how to analyse your results
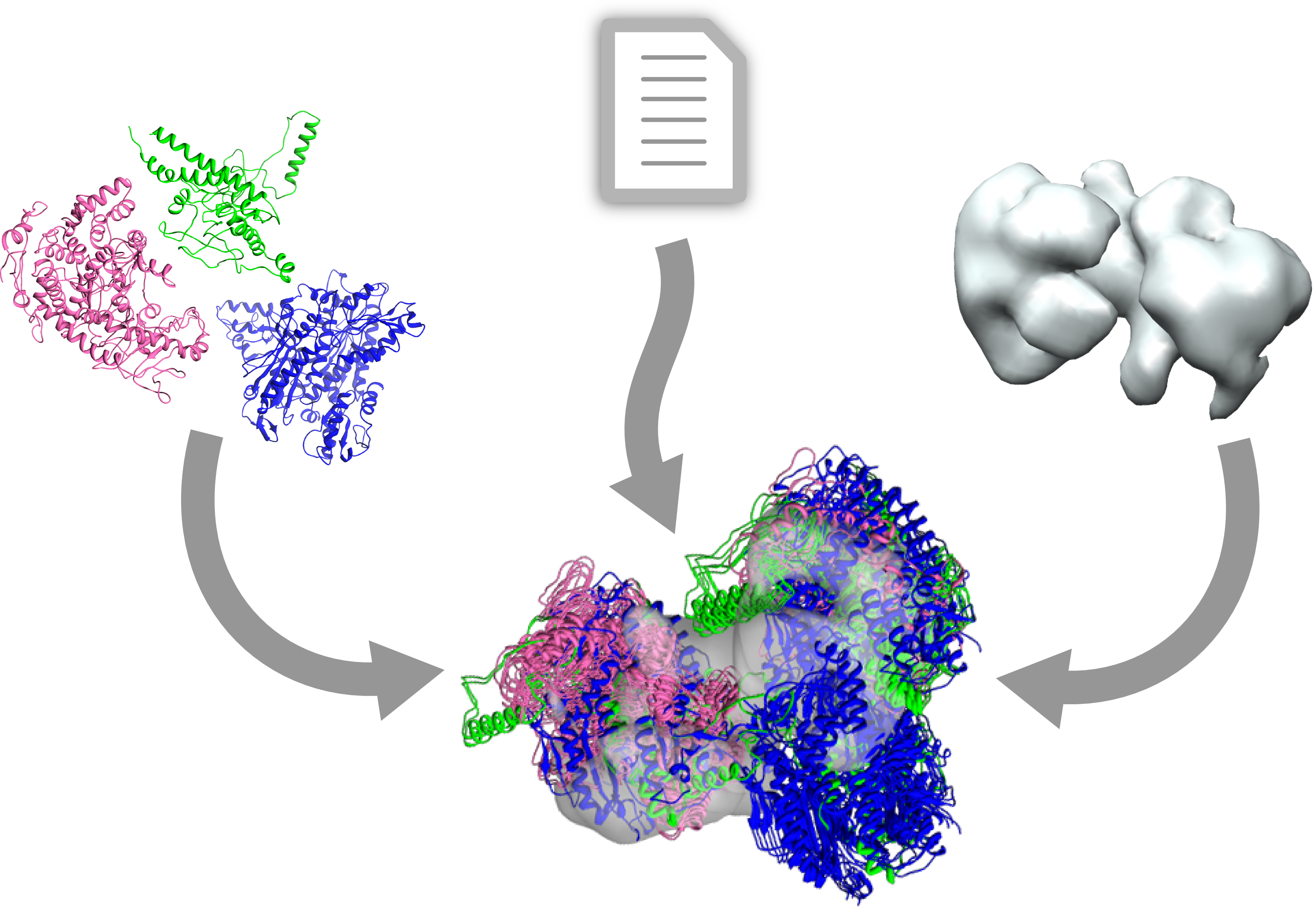
During the tutorial you will build a human polimerase gamma model based on:
- crystal structures of all components
- distance restraints about the system taken from experimental data
- a density map for a whole complex
Biological question we would like to answer is:
- is it possible to build a model of human polimerase gamma complex structure based on gathered data?
Caution!
- Please bear in mind that PyRy3D is a program devoted to low resolution modeling. It is designed to answer a questions, such as whether the available information about a particular complex (from different sources) are sufficient to predict the overall structure of the complex. The obtained models can be later use as a starting points for further higher resolution analysis.
- To analyse large systems built of ten of components, please consider that it will be time consuming and that a large amount of computer power is needed.
Summary:
During the tutorial you will build a model for human polymerase gamma complex. Please bear in mind, that for the modeling process we will use crystal structures of all components, distance restraints taken from analysis of the system. At the end of tutorial you will be able to compare generated model with the „real structure” fit into the density map.
Checking the PyRy3D installation
Instructions on how to install the program can be found in section titled Installing. To be sure that PyRy3D is installed properly on your computer please check the following commands:
If you use the source distribution:
- Start the python interpreter and type:
-
>>> from pyry3d import *
-
If the above command does not return any errors, PyRy3D is ready for usage!
If you use the Windows:
- Open a shell window. Start -> Execute -> cmd
- Go to the directory to which you unpacked PyRy3D. E.g.
-
cd Desktop\PyRy3D
-
- Run the help function of the program:
- >>> from pyry3d import *
If the above command does not return any errors, PyRy3D is ready for usage!
Modeling of human polimerase gamma
- The components' structures
- The components' sequences
- Density map of the complex
- The experimental restraints
- Simulation parameters selection
- Perform simulation
- Results analysis
- Comparison of model with real structure
- Summary and further analysis
Summary
With this example, you will create a model for human polymerase gamma complex. As components we will use the crystal structures of polymerase gamma subdomains:
- pol gamma alfa subdomain
- pol gamma beta subdomain
As a descriptor of the complex shape we will use the density map of the complex (EMDB 1410 )
First please create a working directory where we will run the simulation and store input data for modeling (e.g., polgamma)
We will use the crystal structure of the whole human polimerase gamma complex and divide it into 3 components (chains A, B, C respectively). To do this, please download the crystal structure of the complex from the PDB database. After downloading, divide the single file into three structures: save chains A, B and C in separate pdb files (e.g., A.pdb, B.pdb, C.pdb) and put them into a single folder in your working directory (e.g., polgamma). When the structures are ready, you must put them into tar archive. Such an archive can be used as input data for modeling process [ polgamma.tar ].
The components' sequences
Having set of components' structures prepared now we should make file with sequences. Each component should have its sequence in a FASTA file. The rules are simple:
the definition line should contain the chain name of the particular component as it appears in corresponding PDB file
the sequence should be prepared with only a one letter code
To extract sequences from structures of prepared chains you can use your favourite structure viewer like PyMOL, Chimera or any other tool. To make live easier, we have also prepared an automatic procedure to prepare a FASTA file: for details please visit PyRy3D Chimera Extension site
Having all sequences ready, you should save it in simple .txt file. Example sequence file can be downloaded here [ polgamma.fasta ]
Density map of the complex
Download a density map of the human polymerase gamma complex from EMDB database. Save it in the polgamma folder, untar it and save as emd_1410.map. As you can see, the recommended contour level by the map authors is 0.569
Having all parameters chosen you can save it in simple .txt file. An example map file can be downloaded here [ emd_1410.map ]
The experimental restraints
Investigating a crystal structure of human polymerase gamma complex
Encoding distances as PyRy3D restraints
From literature it is known that some residues are in close proximity to each other: e.g., R232 (polG A domain) and E394 (polG B domain chain C). We have chosen some described interactions and measured the distances between them in the 3IKM structures. The information from those distances can be used as restraints for polymerase complex modeling.
Caution!
- bear in mind that since PyRy3D required the PDB files to be numbered from 1, the numeration of residues in 3IKM structures and prepared components structures might differ
// polymerase gamma distance restraints
dist (
(R162) "A" -(E264) "B" (<=13.5)
(Q446) "A"-(R56) "B" (<=13.00)
(E359-D370) "A" -(R168) "C" (<=13.6)
(E359-D370) "A" - (K284) "C" (<= 11.00)
)
Having all the restraints chosen you should save it in a simple .txt file in abovementioned format. An example of the restraints file can be downloaded here [ restraints.txt ]
Simulation parameters selection
Lets try to model the complex using the simulated annealing procedure. Let's start by setting the annealing temperature to 10 and perform a simulation in 100 steps:
SIMMETHOD SimulatedAnnealing
ANNTEMP 10
STEPS 100
Now lets choose the scoring function weights. As you may know, the scoring function calculates four main parameters: collisions, restraints, empty spaces inside the density map and atoms outside simulation area. Each of these elements has a weight assigned. Thus if you want PyRy3D to focus on a particular aspect of modeling just assign a higher value to its weight. In this example, we are treating all scoring function elements equal so all values stay as default to 1
#scoring function elements
OUTBOX 1 1
MAP_FREESPACE 1 1
CLASHES 1 1
RESTRAINTS 1 1
To define the maximal rotation angle in a single simulation step, use the MAXROT parameter. It helps you to assign the angle value in grades. Similarly, MAXTRANS defines the translation vector values along the x, y, z axes respectively
#move set during simulation
MAXROT 5
MAXTRANS 5 5 5
now the THRESHOLD value which corresponds to the contour level and the SIMBOX value defines how much larger than the density map the simulation area should be. The GRIDRADIUS stands for the radius of a single grid cell, and finally GRIDTYPE can be cubic or diamond
#density map threshold
THRESHOLD 0.6
#grid parameters
SIMBOX 1.6
GRIDRADIUS 1.0
GRIDTYPE X
#simulation scaling process
PARAMSCALINGRANGES 0 25 50
PARAMSCALINGR1 50 100
PARAMSCALINGR2 25 50
PARAMSCALINGR3 0 25
Finally, we can define how often the program should save the results to disk (WRITE_N_ITER)
#pyry output
WRITE_N_ITER 1
Having all the parameters selected, you should save it in simple .txt file. An example of the config file can be downloaded here [ config_file.txt ]
Perform simulation
To run PyRy3D simulation use the following command:
python pyry3d.py -s polgamma.fasta -d polgamma.tar -c config_file.txt -m emd_1410.map -r restraints.txt -o polgamma_out
The ouput files are stored on your disc in polgamma_out folder. The complex models are stored in PDB files. The file name convention is easy: outname_score_iterationNumber.pdb. Apart from the generated pdb files with complex coordinates, PyRy3D also generates a log file where you will find the most important information about simulation process: e.g., the simulation parameters, detailed scores for each saved complex (with final a score, a score for collisions, a score for the restraints, and a score for empty spaces in shape and for atoms outside simulation area)
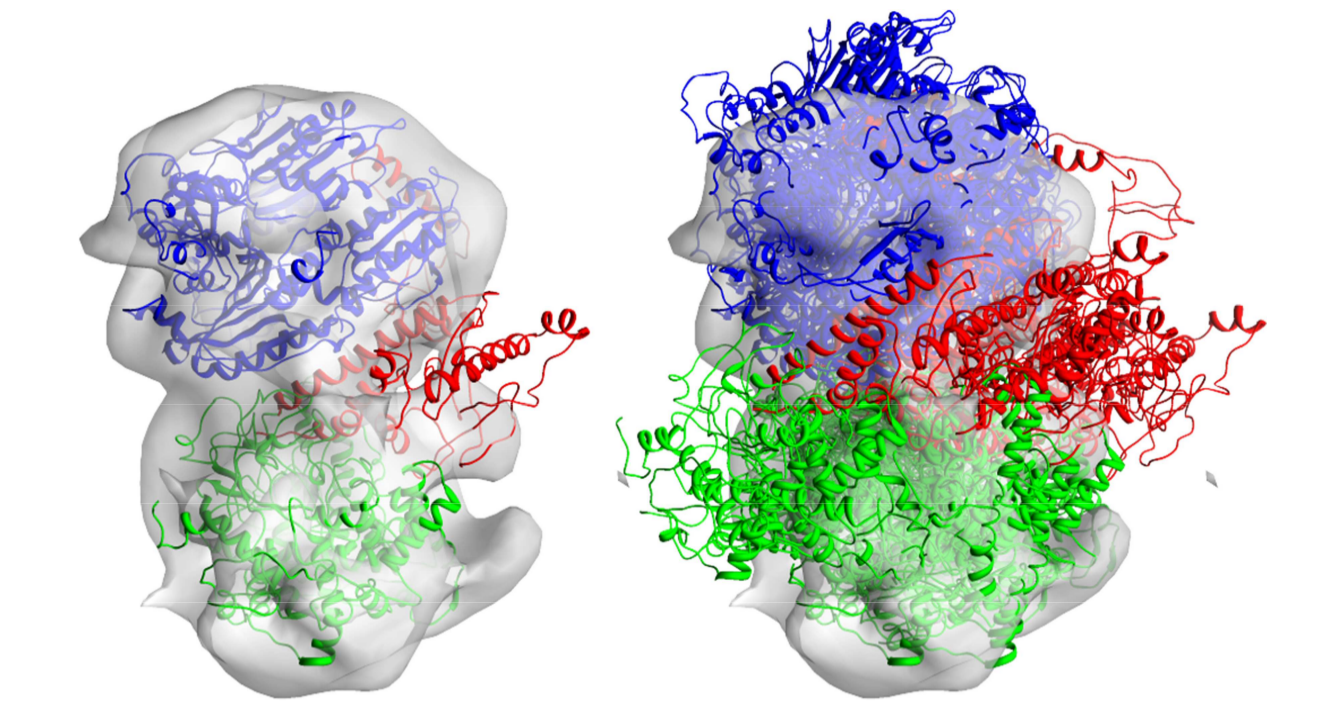
Comparison of model with a real structure
This example involves only a very short simulation that will probably not produce a lot of accurate models of the complex; however, when you run hundreds of simulation steps, you should start to observe their similarities to the native structure. The real human polymerase gamma complex looks like the one shown in the left panel in the picture below. The ensembles of the structures from your PyRy3D run will in some way resemble the structure shown in the right panel.
Having a set of ranked complex models one can:
- cluster the results to find most common results
- choose the best scored models and use other more accurate methods to improve the fit of the into the density map
- analyse the model and publish results :-)