Concepts
- Sequences
- Components Structures
- Experimental restraints
- Simulation parameters
- Complex shape descriptors
- Simulation steps
- Complex scoring function
- Disorder modeling/simulation
Sequences
For all components of a complex program needs information about their sequences. Please provide them following the rules:
- save them in FASTA file
- name each sequence with a chain name as it appears in corresponding structure file
- provide sequence in one-letter code for both proteins or nucleic acids
- sequences for components with undefined structure please name according to the following rule: ChainName_protein for proteins and ChainName_DNA for DNA (_RNA for RNA)
Example:
>A ARTGCVNN >B WRTYPGGH >C SCVNMKYT >X_protein METATTT >Y_DNA AAAAAAAAAA
Components Structures
- crystal structures
PyRy3D can work on crystal structures easily. If you want to include component's crystal structure for modeling just provide PyRy3D with regular .pdb file.
- NMR structures
PyRy3D can work on NMR structures as long as you select one model from an ensemble. If you want to include component's NMR structure for modeling just provide PyRy3D with regular .pdb file with one model inside.
- computational models
PyRy3D can also work on homology/comparative/ab initio models. They are less accurate than crystal and NMR structures but still can provide a lot of information about interactions between components inside a complex. PyRy3D eats .pdb files here as well.
- structures/models with mising fragments
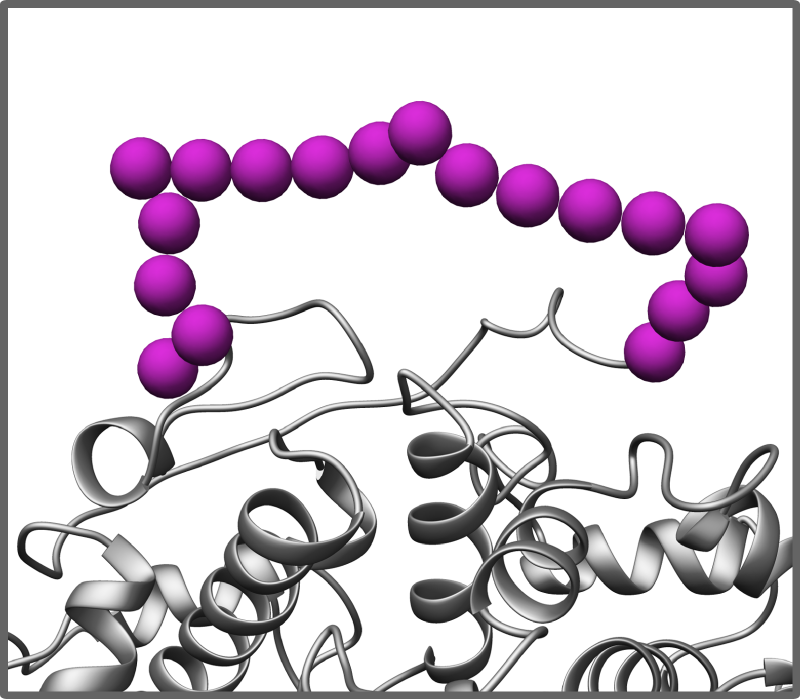
If for some of your components you posess models or experimental structures with missing or disordered fragments, PyRy3D can also work with these. On request missing fragments will be randomly simulated during program run.
- sequences with no structure
For components with no structure you can provide a sequence only and PyRy3D will randomly simulate its volume and will use it as component representation during simulation and complex scoring.
Experimental restraints
If you possess some additional information about your complex e.g. data about interactions between components you can easily incorporate them into modeling procedure. Simply prepare a text file in Filtrest3D format as you can see in the figure below:
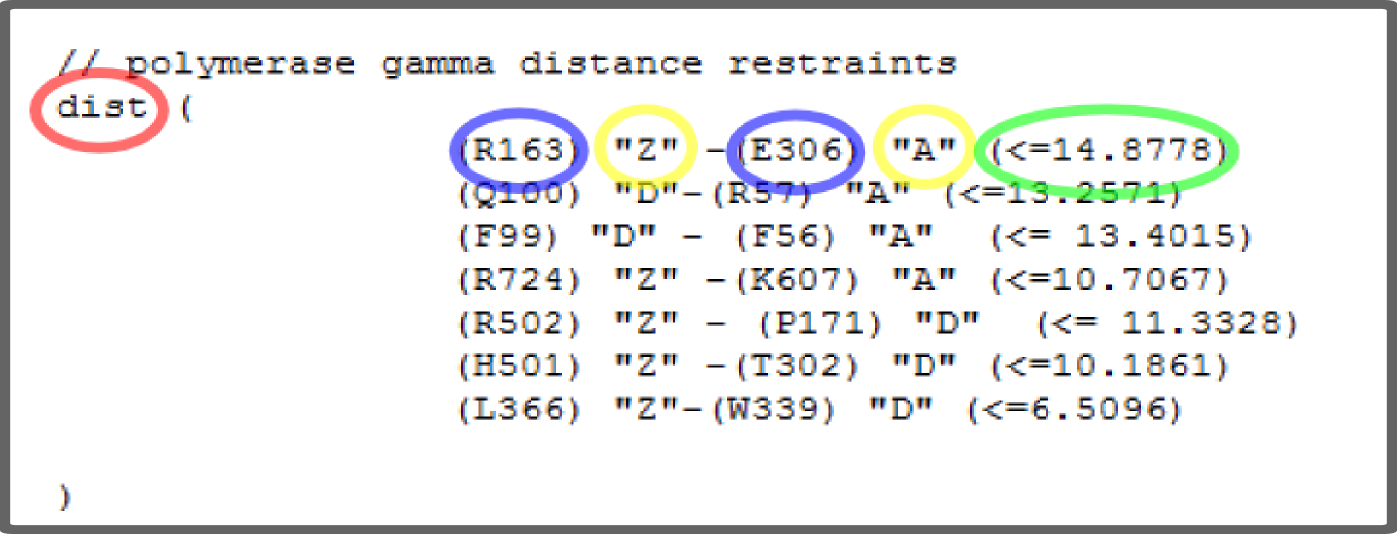
Caution:
// comment starts here and ends at the end of the following line
dist () // "dist" is a keyword to indicate distance as a restraint type, do not forget to put data within "()"
(res1) chain1 - (res2) chain2 (<= dist weight=2)
Where:
- res1 and res2 statnd for name and number of interacting residues
- chain1 and chain2 statnd for names of chains containing corresponding residues resi1 and resi2
- dist is a float value describing distance between resi1 and resi2 in Angstroms
- weight is integer value describing how important is this information for the system
- There are many different possibilities to encode experimental data for details please check Filtrest3D format description
- You can prepare restraints file automatically via Filtrest3D webpage.
Simulation parameters
PyRy3D is a program designed for people who like to have a strong control under modeling process. It enables a user to choose a wide selection of simulation parameters, e.g.:
- technical parameters like number of simulation steps, simulation algorithm, number of outputs etc.
- parameters descibing a target system like electron density threshold describing complex shape, allowed mutations for particular components etc.
- parameters describing scoring function where a user is one who decides on weights of particular components of complex assessment formula
Caution!:
- For detail description about available parameters please check our FAQ
- Having simulation parameters chosen, please save them in a text file and provide as input for PyRy3D
- If you would like to prepare simulation file interactively, please use PyRy3D Chimera Extension which will create a configuration file for you!
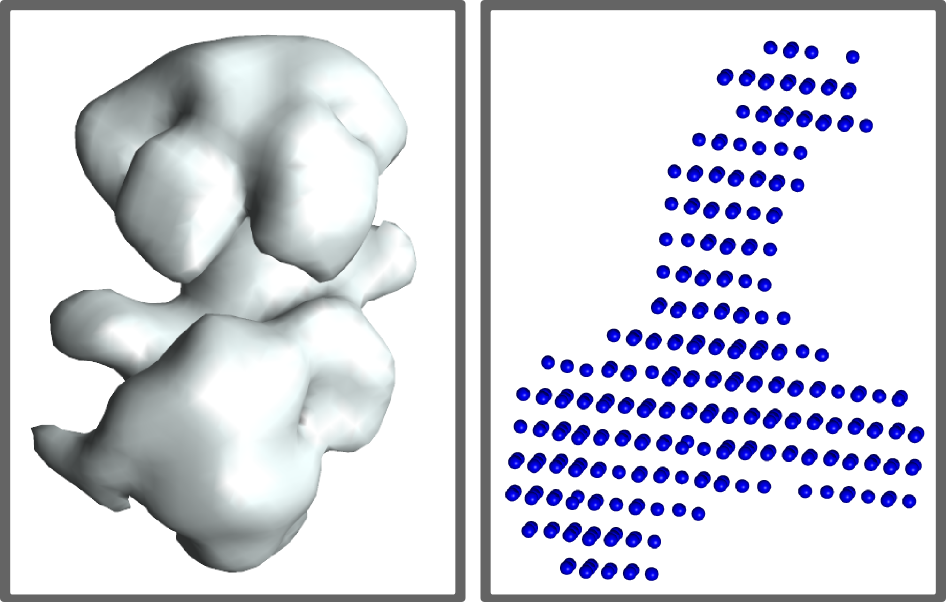
Complex shape descriptors
As a complex shape descriptor PyRy3D can use both: electron density maps in .ccp4 format or ab initio reconstruction models from SAXS experiment.
| Density map |
SAXS ab initio model |
|---|---|
 |
|
Simulation steps
During simulation complex components are mutated. Available possibilities are:
- rotation around X, Y, Z axis
- translation along X, Y, Z axis
- rotation around a line defined by two points in 3D [x1, y1, z1] and [x2,y2,z2]
- two components might be exchanged in 3D space
- disordered regions might be remodelled
- rotation around X, Y, Z axis for all components simultaneously
- translation along X, Y, Z axis for all components simultaneously
After mutation a complex is assessed with scoring function and based on Metropolis condition a generated model is further changed or dismissed from simulatio
Complex scoring function
Each generated complex is validated based to 4 different criteria:
- occurence of collisions between main chains atoms
- violation of distance restraints provided by the user
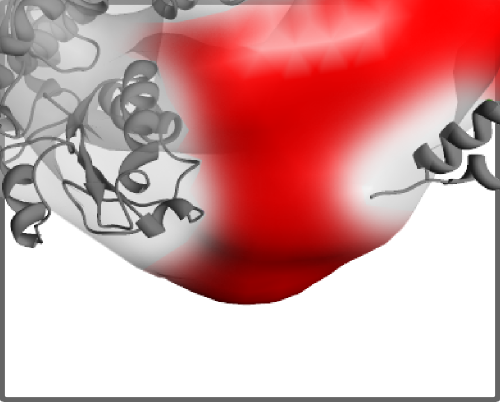
- how well all components are fit into density map considering all density values from cryo-EM map as equal
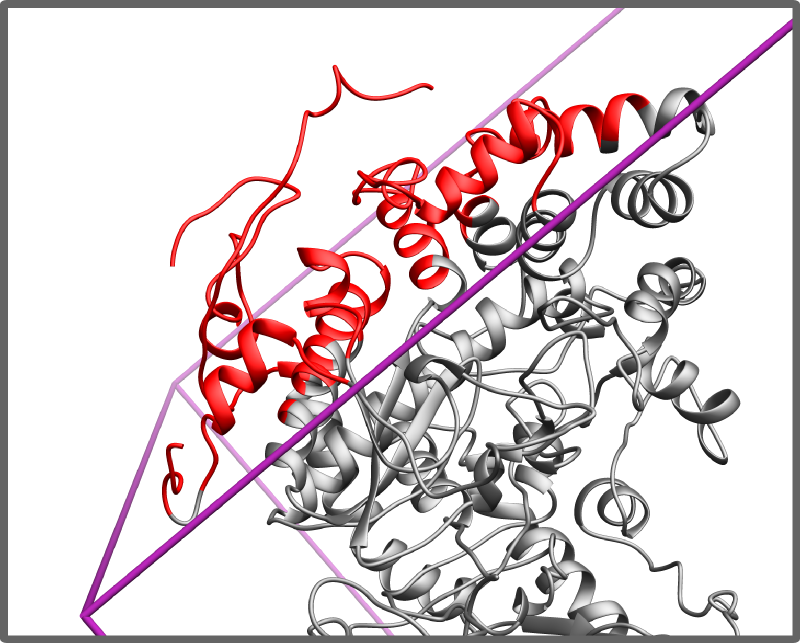
- whether all components are located inside simulation area
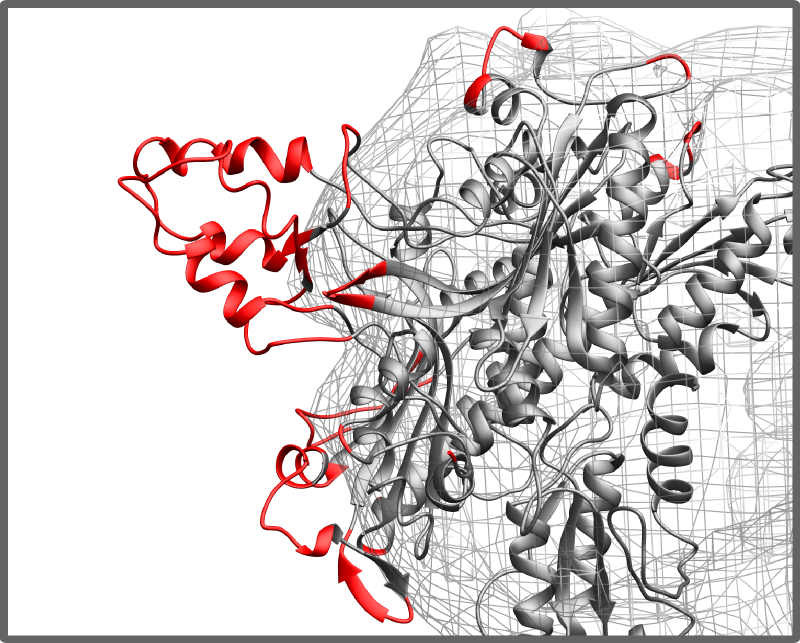
- how well all components are fit into density map considering density values from cryo-EM map
- Score for each complex is calculated based on simple formula:
score = (WEIGHTclashes * clashes_penalty) + (WEIGHTrestraints * restraints_penalty) + (WEIGHTmap_freespace * map_freespace_penalty) + (WEIGHToutbox * outbox_penalty) + (WEIGHTdensity * density_penalty)
where each calculated penalty is multiplied by weight value provided by the user
- Collision detection and penalties for collided atoms/residues
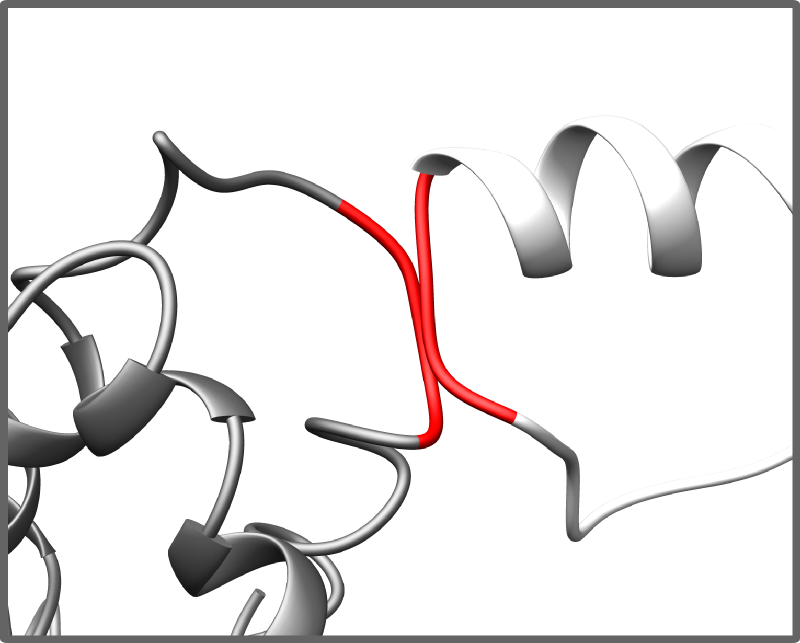
Restraints validation and penalties for violation of restraints
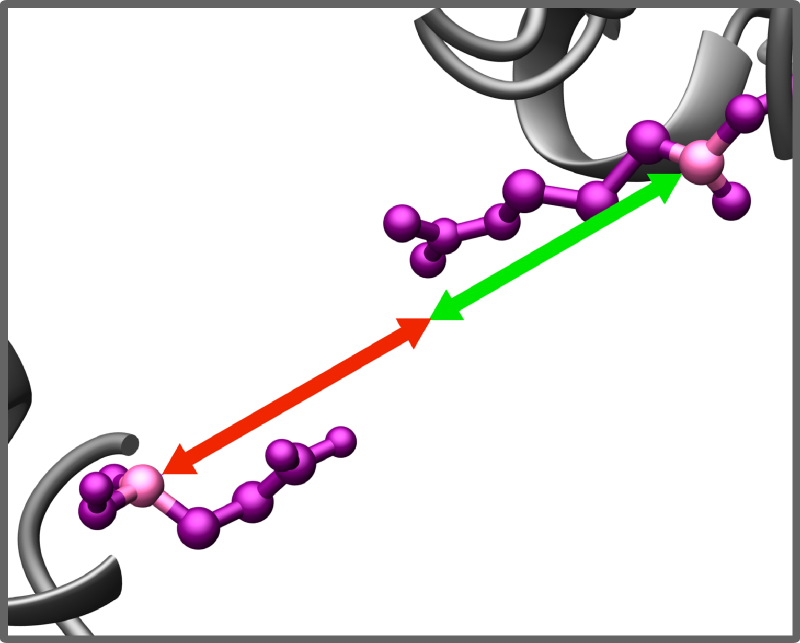
Detecting atoms/residues outside the simulation are
Detecting empty spaces inside a density map
Detecting empty spaces for atoms/residues outside a density map
Disorder modeling/simulation
Unique feature of PyRy3D program is its ability to model regions of components with unassigned structure. For these fragments PyRy3D randomly chooses positions of atoms in gas-like behaviour. Such procedure enables to model systems where some framgnets of components are missing.