mqapRNA
Exemplary output
The exemplary output for the demo input can be found at/mqapRNA/jobs/62d2e3ea-f7b4-4e56-b3f1-13f84c84c110
Input
RNA structure - mqapRNA accepts input files in the following formats: (1) (old and new) PDB format with a single model or multi-models files (2) a zip file with a set of PDB files. If you use a zip file, please make sure that there are not additional folders in the zip file. A zip should look as:rp17.zip file1.pdb file2.pdb
Up to 100 structures is allowed to score with mqapRNA.
Only lines starting with
ATOM are taken under consideration! If any heteroatoms should be taken under consideration during the docking process, change word HETATM to ATOM in the pdb file.
If you have any problem with preparation of your file, let us know mqapRNA@genesilico.pl
Job title – could be any string.
E-mail address – the email address will be used to send results, but is not required of the user. However, for long sequences, the simulation can take many hours, so if the page is lost, there is no way to find it again.
RNA secondary structure [optional]
RNA secondary structure, e.g.,
((((.[[[[[[.))))........((((.....]]]]]]...(((((....)))))..))))
. represents an unpaired nucleotide, ( ) represents a base pair, [ ] represents base pair of pseudoknots
For secondary structure prediction with the use of SHAPE data, RNA structure (5.8.1; Jun 16, 2016) is used. The format required for a SHAPE data file is as following https://rna.urmc.rochester.edu/Text/File_Formats.html#SHAPE
FOR NOW, mqapRNA CAN SCORE ONLY SINGLE CHAIN STRUCTURES.
Secondary structure comparisons were calculated based on outputs of ClaRNA (Waleń et al. 2014) using the Interaction Network Fidelity (INF) value which is computed as:
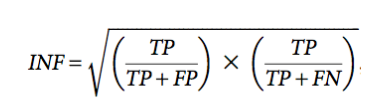
where TP is the number of correctly predicted base–base interactions, FP is the number of predicted base–base interactions with no correspondence in the solution model, and FN is the number of base–base interactions in the solution model not present in the predicted model (Miao et al. 2017).
To calculate penalty, a calculated INF between a secondary structure of a model and a submitted secondary structure is subtracted from 1. In that way, a wrong secondary structure (an INF closer to 0) gives a high penalty (closer to 1).
Distance restraints definition [optional]
The Format for distance restraints:
d:[chain id][resiude index]-[chain id][residue index] x [distance] # where x can be >, <, =for example:
d:A14-A60 < 12which means, residue 14 of the chain A and residue 60 of the chain A should be in distance below 12 Å.
For evolutionary restraints (direct coupling analysis, DCA) (Weinreb et al. 2016), the suggested distance is 7 Å, while for MOHCA-seq, 25 Å (Das et al. 2008). Analysis of those scores can help the user to decide which structure to select for further investigation.
Das R, Kudaravalli M, Jonikas M, Laederach A, Fong R, Schwans JP, et al. Structural inference of native and partially folded RNA by high-throughput contact mapping. Proceedings of the National Academy of Sciences. National Acad Sciences; 2008 Mar 18;105(11):4144–9.
Miao Z, Westhof E. RNA Structure: Advances and Assessment of 3D Structure Prediction. Annu Rev Biophys. 2017 May 22;46(1):483–503.
Waleń T, Chojnowski G, Gierski P, Bujnicki JM. ClaRNA: a classifier of contacts in RNA 3D structures based on a comparative analysis of various classification schemes. Nucleic Acids Res. Oxford University Press; 2014 Oct 29;42(19):e151–1.
Weinreb C, Riesselman AJ, Ingraham JB, Gross T, Sander C, Marks DS. 3D RNA and Functional Interactions from Evolutionary Couplings. Cell. Elsevier Inc; 2016 Oct 1;:1–14.